Bệnh loạn dưỡng bạch cầu dị sắc là gì?
Bệnh loạn dưỡng bạch cầu dị sắc (Metachromatic Leukodystrophy – MLD) là một bệnh lý di truyền hiếm gặp, gây tổn thương tiến triển chất trắng của hệ thần kinh trung ương (não và tủy sống), đồng thời ảnh hưởng đến các dây thần kinh ngoại biên. Bệnh thuộc nhóm rối loạn chuyển hóa lysosome, trong đó cơ thể không thể phân hủy bình thường một số chất béo đặc hiệu trong tế bào thần kinh.
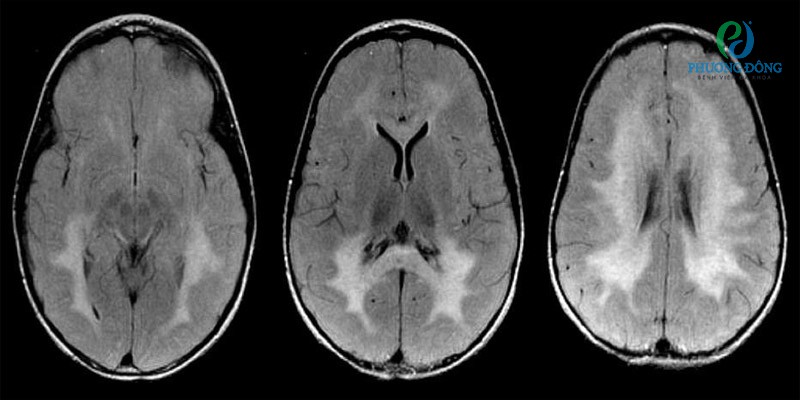 Hình ảnh cộng hưởng từ (MRI) mặt cắt ngang Flair
Hình ảnh cộng hưởng từ (MRI) mặt cắt ngang Flair
Ghi chú:
- Lưu ý các tổn thương chất trắng lan rộng hai bên, nổi bật hơn ở vùng phía sau.
- Chẩn đoán: Một bé gái 3 tuổi có các triệu chứng co cứng cơ tiến triển, suy giảm thị lực và chậm phát triển vận động.
Sự rối loạn này dẫn đến thoái hóa myelin – lớp vỏ bảo vệ sợi thần kinh, khiến quá trình dẫn truyền tín hiệu thần kinh bị gián đoạn. Hậu quả là người bệnh xuất hiện suy giảm dần chức năng vận động và nhận thức, với các biểu hiện ngày càng nặng theo thời gian. Phần lớn các trường hợp MLD có tiên lượng xấu và có thể dẫn đến tử vong sau nhiều năm kể từ khi được chẩn đoán.
Tên gọi “loạn dưỡng bạch cầu dị sắc” bắt nguồn từ đặc điểm mô bệnh học: các tế bào thần kinh tích tụ sulfatide – một loại lipid bất thường. Khi quan sát dưới kính hiển vi và nhuộm màu đặc hiệu, các sulfatide này bắt màu khác biệt so với mô xung quanh (hiện tượng “dị sắc”), từ đó hình thành thuật ngữ metachromatic.
Về bản chất, loạn dưỡng bạch cầu dị sắc là quá trình phá hủy tiến triển của chất trắng, làm suy giảm chức năng thần kinh không hồi phục nếu không được phát hiện và can thiệp sớm.
Các dạng bệnh loạn dưỡng bạch cầu dị sắc
Bệnh loạn dưỡng bạch cầu dị sắc được phân loại thành ba thể lâm sàng, dựa trên độ tuổi khởi phát và tốc độ tiến triển của bệnh:
Thể muộn ở trẻ sơ sinh: Thể bệnh này thường khởi phát ở trẻ từ 12–20 tháng tuổi. Trẻ bắt đầu xuất hiện các rối loạn về đi lại, chậm phát triển vận động, sau đó tiến triển nhanh sang mù lòa, sa sút trí tuệ và mất dần khả năng vận động. Đây là thể bệnh nặng và tiến triển nhanh nhất, với nguy cơ tử vong cao, đa số trẻ không sống quá 5 tuổi. Thể muộn ở trẻ sơ sinh chiếm khoảng 50–60% tổng số ca MLD.
Thể thiếu niên: Thể thiếu niên thường xuất hiện ở trẻ trong độ tuổi 3–10 tuổi. Biểu hiện ban đầu thường là suy giảm khả năng học tập, thay đổi hành vi, sau đó xuất hiện co giật, rối loạn vận động và sa sút trí tuệ. Bệnh tiến triển chậm hơn so với thể nhũ nhi, nhưng vẫn dẫn đến tử vong sau 10–20 năm kể từ thời điểm chẩn đoán. Thể này chiếm khoảng 20–30% số trường hợp.
Thể khởi phát ở người lớn: Thể người lớn thường bắt đầu sau 16 tuổi, đôi khi bị chẩn đoán nhầm với các rối loạn tâm thần. Triệu chứng đặc trưng gồm thay đổi tính cách, rối loạn hành vi, sa sút trí tuệ, co giật và suy giảm vận động. Tiến triển bệnh chậm hơn nhưng vẫn dẫn đến tử vong trong khoảng 6–14 năm sau chẩn đoán. Thể này chiếm khoảng 15–20% các ca MLD.
Các biểu hiện của bệnh loạn dưỡng bạch cầu dị sắc
Biểu hiện của bệnh loạn dưỡng bạch cầu dị sắc thay đổi tùy theo thể bệnh và độ tuổi khởi phát. Tuy nhiên, điểm chung của tất cả các thể là sự suy thoái tiến triển của hệ thần kinh, ảnh hưởng đến vận động cơ bắp, khả năng nhận thức, hành vi, cảm xúc và nhân cách. Khi bệnh tiến triển, người bệnh dần mất khả năng tự chủ trong sinh hoạt hằng ngày.
Ở trẻ sơ sinh
 Trẻ có thể gặp khó khăn trong việc ăn uống, nôn mửa hoặc khi đứng và đi lại
Trẻ có thể gặp khó khăn trong việc ăn uống, nôn mửa hoặc khi đứng và đi lại
Trẻ mắc loạn dưỡng bạch cầu dị sắc thể khởi phát sớm thường phát triển bình thường trong những tháng đầu đời. Tuy nhiên, từ khoảng 9–12 tháng tuổi, các dấu hiệu thần kinh bắt đầu xuất hiện và tiến triển nhanh chóng, bao gồm:
- Khó khăn khi đứng và đi lại, sau đó mất hoàn toàn khả năng vận động;
- Giảm trương lực cơ, cơ mềm nhão;
- Chậm phát triển vận động và nhận thức;
- Rối loạn ngôn ngữ, khó phát âm;
- Suy giảm thị lực tiến triển, có thể dẫn đến mù lòa;
- Khó nuốt, tăng nguy cơ sặc và viêm phổi hít;
- Sa sút trí tuệ tiến triển.
Thể bệnh này thường diễn tiến nặng và có tiên lượng kém nếu không được can thiệp sớm.
Ở trẻ vị thành niên
Ở nhóm tuổi vị thành niên, bệnh thường khởi phát âm thầm hơn nhưng tác động rõ rệt đến học tập và hành vi xã hội. Các triệu chứng thường gặp gồm:
- Suy giảm trí tuệ, biểu hiện bằng kết quả học tập sa sút;
- Rối loạn hành vi, khó thích nghi xã hội;
- Thay đổi tính cách rõ rệt;
- Rối loạn vận động, giảm khả năng kiểm soát cơ bắp;
- Bệnh lý thần kinh ngoại biên gây tê bì, yếu chi;
- Xuất hiện các cơn động kinh;
- Sa sút trí tuệ tiến triển theo thời gian.
Ở người lớn
Loạn dưỡng bạch cầu dị sắc khởi phát ở người lớn thường biểu hiện chủ yếu bằng rối loạn tâm thần, trong khi triệu chứng vận động có thể nhẹ hoặc xuất hiện muộn. Những dấu hiệu ban đầu thường dễ bị nhầm lẫn với các rối loạn tâm lý – tâm thần khác, như khó khăn trong học tập, công việc hoặc thay đổi hành vi xã hội.
Các triệu chứng điển hình bao gồm:
- Rối loạn tâm thần như hoang tưởng, ảo giác, rối loạn loạn thần hoặc biểu hiện giống tâm thần phân liệt;
- Co giật;
- Tổn thương thần kinh ngoại biên;
- Sa sút trí tuệ tiến triển.
Xem thêm:
Khi nào người bệnh cần gặp bác sĩ?
Hãy trao đổi với bác sĩ nếu bạn nhận thấy bất kỳ dấu hiệu nào được liệt kê phía trên hoặc nếu bạn lo ngại về các dấu hiệu hoặc triệu chứng của chính mình.
Nguyên nhân gây bệnh loạn dưỡng bạch cầu dị sắc
Loạn dưỡng bạch cầu dị sắc là bệnh di truyền lặn trên nhiễm sắc thể thường, xảy ra khi người bệnh nhận gen đột biến từ cả cha và mẹ.
Phần lớn các trường hợp bệnh liên quan đến đột biến gen ARSA– gen chịu trách nhiệm mã hóa enzyme arylsulfatase A. Enzyme này có vai trò phân giải sulfatide, một thành phần lipid quan trọng trong bao myelin của hệ thần kinh. Khi enzyme bị thiếu hụt hoặc hoạt động kém, sulfatide tích tụ bất thường, gây độc cho chất trắng, làm tổn thương tế bào thần kinh và dẫn đến thoái hóa thần kinh tiến triển.
Ngoài ra, một số trường hợp hiếm gặp liên quan đến đột biến gen PSAP, gen này cũng tham gia vào quá trình chuyển hóa sulfatide. Khi gen PSAP bị rối loạn, cơ chế phân giải sulfatide bị gián đoạn, góp phần thúc đẩy quá trình tổn thương hệ thần kinh tương tự như đột biến ARSA.
Tại sao bệnh loạn dưỡng bạch cầu dị sắc thường bị phát hiện muộn?
Bệnh loạn dưỡng bạch cầu dị sắc thường được chẩn đoán ở giai đoạn muộn do triệu chứng khởi phát không đặc hiệu, tiến triển âm thầm và dễ bị nhầm lẫn với các rối loạn phát triển thần kinh khác. Điều này khiến bác sĩ và gia đình khó nghĩ tới bệnh ở giai đoạn sớm, dẫn đến việc chỉ định xét nghiệm chuyên sâu bị trì hoãn.
Ở giai đoạn đầu, trẻ có thể chỉ biểu hiện chậm phát triển vận động, khó đi lại, chậm nói, sa sút khả năng học tập hoặc xuất hiện thay đổi hành vi nhẹ. Những dấu hiệu này thường không điển hình và dễ bị quy kết là chậm phát triển thông thường hoặc rối loạn tâm lý – hành vi.
Đặc biệt, ở thể sơ sinh muộn và thể thiếu niên, bệnh tiến triển rất chậm, có thể kéo dài nhiều tháng đến vài năm trước khi xuất hiện các triệu chứng thần kinh rõ rệt như mất khả năng vận động, co cứng cơ hoặc suy giảm nhận thức nặng. Khi đó, tổn thương chất trắng não đã lan rộng và bệnh thường đã ở giai đoạn muộn, làm giảm hiệu quả điều trị.
Bên cạnh đó, loạn dưỡng bạch cầu dị sắc là bệnh hiếm gặp, tỷ lệ mắc thấp khiến mức độ nhận biết trong cộng đồng và hệ thống y tế còn hạn chế. Đây cũng là nguyên nhân khiến nhiều bệnh nhân bỏ lỡ “thời điểm vàng” can thiệp, đặc biệt với các phương pháp điều trị nhằm làm chậm tiến triển bệnh.
Biến chứng của bệnh loạn dưỡng bạch cầu dị sắc
Khi bệnh tiến triển, người bệnh có thể đối mặt với nhiều biến chứng nghiêm trọng, bao gồm:
- Suy giảm nhận thức và chức năng thần kinh, tiến triển thành sa sút trí tuệ;
- Mù lòa do teo dây thần kinh thị giác;
- Suy dinh dưỡng do rối loạn nuốt và giảm vận động;
- Viêm phổi hít, xuất phát từ sặc thức ăn hoặc dịch tiết đường hô hấp;
- Tử vong, thường do nhiễm trùng, suy hô hấp hoặc biến chứng toàn thân.
Các lựa chọn điều trị bệnh loạn dưỡng bạch cầu dị sắc
Hiện nay, chưa có phương pháp điều trị khỏi hoàn toàn bệnh loạn dưỡng bạch cầu dị sắc. Mục tiêu điều trị chủ yếu là làm chậm tiến triển bệnh, kiểm soát triệu chứng và cải thiện chất lượng cuộc sống cho người bệnh.
Ở trẻ chưa có triệu chứng hoặc mới xuất hiện triệu chứng nhẹ, một số chuyên gia có thể cân nhắc ghép tế bào gốc tạo máu, nhằm làm chậm tổn thương thần kinh nếu được thực hiện sớm.
Điều trị hỗ trợ bằng thuốc
Điều trị triệu chứng thường bao gồm các nhóm thuốc sau:
- Thuốc chống co giật: kiểm soát động kinh;
- Thuốc giãn cơ: giảm co cứng và co thắt cơ;
- Thuốc điều chỉnh tâm trạng: hỗ trợ rối loạn cảm xúc, trầm cảm;
- Thuốc giảm đau: chủ yếu là thuốc chống viêm không steroid và các thuốc giảm đau phù hợp khác.
Các liệu pháp hỗ trợ chức năng
 Phục hồi chức năng dựa trên tình trạng sức khỏe cụ thể của từng bệnh nhân và sự tư vấn của các chuyên gia y tế
Phục hồi chức năng dựa trên tình trạng sức khỏe cụ thể của từng bệnh nhân và sự tư vấn của các chuyên gia y tế
Ngoài điều trị bằng thuốc, người bệnh thường cần phối hợp nhiều liệu pháp chuyên biệt:
- Vật lý trị liệu giúp duy trì sức mạnh cơ bắp và hạn chế cứng khớp;
- Phục hồi chức năng nhằm hỗ trợ khả năng tự chăm sóc và sinh hoạt hằng ngày;
- Liệu pháp ngôn ngữ giúp cải thiện các vấn đề về nói và nuốt;
- Liệu pháp tâm lý hỗ trợ sức khỏe tinh thần cho người bệnh và gia đình;
- Mở thông dạ dày qua da bằng nội soi (PEG) khi người bệnh gặp khó khăn nghiêm trọng trong ăn uống và đảm bảo dinh dưỡng.
Trong những năm gần đây, liệu pháp gen và các phương pháp điều trị sinh học đang mở ra nhiều hy vọng mới cho bệnh loạn dưỡng bạch cầu dị sắc. Liệu pháp gen hướng tới can thiệp trực tiếp vào nguyên nhân gốc rễ của bệnh, bằng cách sửa chữa hoặc thay thế gen bị đột biến. Tuy nhiên, các phương pháp này hiện vẫn đang trong giai đoạn nghiên cứu và thử nghiệm lâm sàng, chưa được áp dụng rộng rãi.
Kết luận
Bệnh loạn dưỡng bạch cầu dị sắc là một bệnh lý tiến triển, không thể tự hồi phục và có thể để lại hậu quả nặng nề nếu không được phát hiện kịp thời. Việc hiểu đúng về các dấu hiệu sớm, nguy cơ di truyền và hướng tiếp cận chẩn đoán giúp gia đình và người chăm sóc chủ động hơn trong việc thăm khám, theo dõi và can thiệp sớm. Trong bối cảnh y học hiện đại ngày càng có nhiều tiến bộ về điều trị hỗ trợ và liệu pháp chuyên sâu, phát hiện bệnh ở giai đoạn sớm chính là cơ hội quan trọng nhất để cải thiện tiên lượng và kéo dài chất lượng sống cho người bệnh.






