Loạn dưỡng cơ Duchenne là một dạng bệnh di truyền nghiêm trọng khiến cơ bắp yếu dần theo thời gian, thường khởi phát sớm ở trẻ nhỏ và tiến triển nhanh nếu không được can thiệp đúng cách. Dù hiện nay chưa có phương pháp chữa khỏi hoàn toàn, nhưng y học đã ghi nhận nhiều tiến bộ trong điều trị giúp làm chậm quá trình thoái hoá cơ, cải thiện khả năng vận động và nâng cao chất lượng sống cho người bệnh.
Bệnh loạn dưỡng cơ Duchenne là gì?
Loạn dưỡng cơ Duchenne (Duchenne Muscular Dystrophy – DMD) là một bệnh lý di truyền tiến triển, đặc trưng bởi tình trạng các sợi cơ bị yếu dần và thoái hóa theo thời gian. Bệnh chủ yếu ảnh hưởng đến nhóm cơ vận động, cơ hô hấp và cơ tim, khiến người bệnh dần mất khả năng đi lại, hô hấp suy yếu và tăng nguy cơ biến chứng tim mạch.
DMD là rối loạn liên quan đến đột biến gene DMD, dẫn đến thiếu hụt dystrophin – một protein thiết yếu giúp ổn định cấu trúc sợi cơ. Khi dystrophin không được sản xuất đầy đủ, các tế bào cơ dễ tổn thương, thoái hóa và bị thay thế bằng mô mỡ hoặc mô xơ.
Bệnh mang tính di truyền lặn liên kết nhiễm sắc thể X, vì vậy nam giới là nhóm bị ảnh hưởng nhiều nhất. Các dấu hiệu thường xuất hiện từ 3– 5 tuổi, tiến triển nhanh và nặng dần qua các năm.
 Trẻ gặp khó khăn khi đi lại, vận động khi mắc hội chứng loạn dưỡng cơ Duchenne
Trẻ gặp khó khăn khi đi lại, vận động khi mắc hội chứng loạn dưỡng cơ Duchenne
Theo thống kê, loạn dưỡng cơ Duchenne xảy ra ở khoảng 1/3.600 trẻ sơ sinh nam, trở thành dạng loạn dưỡng cơ phổ biến nhất và nghiêm trọng nhất hiện nay. Dù chưa có phương pháp chữa khỏi hoàn toàn, nhiều liệu pháp mới như corticosteroid, thuốc nhắm trúng đích và các biện pháp hỗ trợ vận động – hô hấp có thể giúp làm chậm tiến triển bệnh, giảm biến chứng và cải thiện chất lượng cuộc sống.
Đối tượng có nguy cơ mắc loạn dưỡng cơ Duchenne
Loạn dưỡng cơ Duchenne có thể xuất hiện ở mọi chủng tộc và cả hai giới. Tuy nhiên, do cơ chế di truyền liên kết nhiễm sắc thể X, bệnh gặp chủ yếu ở bé trai, trong khi bé gái thường là người mang gene bệnh (carrier) và có thể truyền gene đột biến cho thế hệ sau.
Những đối tượng có nguy cơ cao bao gồm:
- Trẻ trai sinh ra từ mẹ mang gene DMD đột biến.
- Người có tiền sử gia đình mắc loạn dưỡng cơ (đặc biệt là họ hàng gần: anh, em, cậu, dì).
- Bé gái mang gene đột biến: thường không biểu hiện bệnh nặng nhưng có khả năng truyền gene sang con.
Việc tầm soát gene và tư vấn di truyền trước sinh đóng vai trò quan trọng trong phát hiện sớm nguy cơ mắc bệnh.
Nguyên nhân gây ra chứng loạn dưỡng cơ Duchenne
Loạn dưỡng cơ Duchenne xuất phát từ một biến thể (đột biến) trên gen DMD, gen này có nhiệm vụ hướng dẫn cơ thể tạo ra protein dystrophin. Đây là loại protein giữ vai trò quan trọng trong việc duy trì cấu trúc, độ bền và khả năng phục hồi của sợi cơ khi vận động.
Khi trẻ mắc DMD, cơ thể không sản xuất hoặc chỉ sản xuất được lượng dystrophin rất nhỏ, không đủ để bảo vệ tế bào cơ. Do thiếu dystrophin, các sợi cơ trở nên yếu và dễ tổn thương, dẫn đến quá trình thoái hóa – hoại tử tế bào cơ diễn ra liên tục. Theo thời gian, các tế bào cơ bị phá huỷ không thể tái tạo như bình thường, và phần khoảng trống này được thay thế bằng mỡ và mô sẹo, khiến cho việc vận động ngày càng khó khăn hơn.
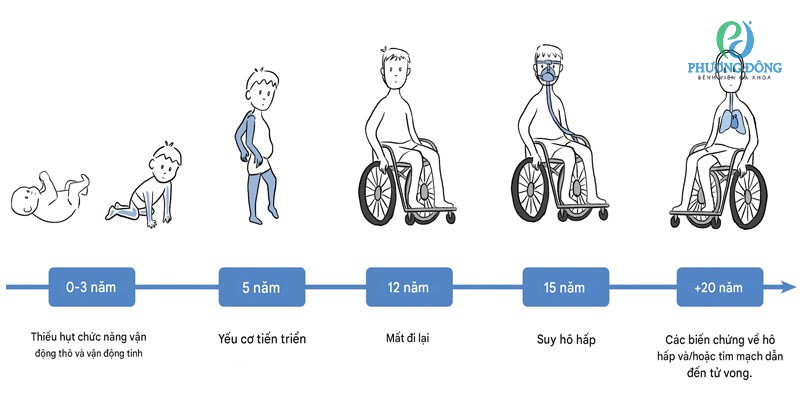 Sơ đồ mô tả diễn tiến lâm sàng của bệnh loạn dưỡng cơ Duchenne bao gồm độ tuổi, các vùng cơ thể bị ảnh hưởng và các giai đoạn bệnh
Sơ đồ mô tả diễn tiến lâm sàng của bệnh loạn dưỡng cơ Duchenne bao gồm độ tuổi, các vùng cơ thể bị ảnh hưởng và các giai đoạn bệnh
Cơ chế di truyền
Loạn dưỡng cơ Duchenne là bệnh di truyền lặn liên kết nhiễm sắc thể X, tức là gen gây bệnh nằm trên nhiễm sắc thể X – một trong hai nhiễm sắc thể quy định giới tính.
Cách di truyền hoạt động như sau:
- Gen DMD nằm trên nhiễm sắc thể X.
- Bé trai có cặp nhiễm sắc thể XY (X từ mẹ, Y từ bố).
- Bé gái có cặp nhiễm sắc thể XX (mỗi X từ bố và mẹ).
Vì bé trai chỉ có một nhiễm sắc thể X, nên nếu nhiễm sắc thể X đó mang đột biến gây DMD, trẻ chắc chắn mắc bệnh do không có bản sao X còn lại để bù trừ.
Ngược lại, bé gái có hai nhiễm sắc thể X, khi mang một bản sao gen bị đột biến, nhiễm sắc thể X còn lại có thể bù đắp. Vì vậy:
- Bé gái thường không biểu hiện triệu chứng hoặc triệu chứng rất nhẹ.
- Bé gái có thể trở thành người mang gen (carrier).
- Người mang gen vẫn có khả năng truyền đột biến cho con trong các lần mang thai tiếp theo.
Khoảng 30% (3/10) trường hợp DMD không phải do di truyền mà do đột biến gen xảy ra ngẫu nhiên trong quá trình hình thành phôi. Điều này đồng nghĩa gia đình không có tiền sử bệnh, và sự thay đổi gen xảy ra một cách tự phát.
Hiện nay, không có biện pháp nào ngăn ngừa loạn dưỡng cơ Duchenne, nhưng xét nghiệm di truyền và tư vấn di truyền có thể giúp các gia đình đánh giá nguy cơ trong tương lai.
Xem thêm:
Các triệu chứng của hội chứng loạn dưỡng cơ Duchenne
Các biểu hiện lâm sàng của loạn dưỡng cơ Duchenne (DMD) thường tiến triển âm thầm nhưng ngày càng rõ rệt theo thời gian. Những triệu chứng điển hình bao gồm:
- Giảm khả năng vận động và kém linh hoạt trong các hoạt động thường ngày;
- Yếu cơ vùng gốc chi (vai, hông), khiến việc đứng lên hoặc leo cầu thang trở nên khó khăn;
- Teo cơ tại nhiều vị trí, đặc biệt ở các nhóm cơ lớn;
- Bắp chân phì đại đối xứng hai bên do tình trạng giả phì đại cơ;
- Nồng độ men Creatine Kinase (CK) trong máu tăng rất cao, có thể gấp hơn 20 lần so với giới hạn bình thường, cho thấy mức độ tổn thương cơ nghiêm trọng.
Các giai đoạn tiến triển của bệnh
DMD thường tiến triển qua nhiều mức độ từ nhẹ đến nặng, được mô tả rõ như sau:
- Giai đoạn nhẹ: Trẻ dễ mệt, yếu cơ nhẹ, di chuyển khó khăn, hay vấp ngã, dáng đi bất thường, chạy chậm, đi nhón chân. Giai đoạn này chưa xuất hiện dấu hiệu Gowers.
- Giai đoạn trung bình: Xuất hiện dấu hiệu Gowers dương tính, trẻ khó leo cầu thang và dáng đi lạch bạch do cơ gốc chi yếu dần.
- Giai đoạn nặng: Trẻ không thể tự đứng lên nếu không có người hỗ trợ, khả năng đi lại giảm rõ rệt, teo cơ tiến triển mạnh.
- Giai đoạn cuối: Mất hoàn toàn khả năng vận động và phải sử dụng xe lăn để di chuyển.
Biến chứng nguy hiểm khi mắc loạn dưỡng cơ Duchenne
DMD không chỉ ảnh hưởng đến cơ xương mà còn tác động đến nhiều cơ quan khác. Nếu không được theo dõi và điều trị, bệnh có thể gây ra những biến chứng nghiêm trọng như:
- Bệnh cơ tim và giảm chức năng tim;
- Biến dạng chi, đặc biệt là ở tay hoặc chân;
- Suy tim sung huyết (ít gặp nhưng có thể xảy ra ở giai đoạn muộn);
- Rối loạn nhịp tim (hiếm gặp nhưng tiềm ẩn nguy cơ cao);
- Suy nhược cơ thể, stress kéo dài và ảnh hưởng tâm lý;
- Viêm phổi hoặc các nhiễm trùng hô hấp do yếu cơ hô hấp;
- Mất khả năng vận động, suy giảm hoàn toàn khả năng tự chăm sóc.
Khi nào cần gặp bác sĩ?
Bạn nên liên hệ với bác sĩ khi xuất hiện một trong các dấu hiệu sau để được đánh giá và điều trị kịp thời:
- Các biểu hiện nghi ngờ giống loạn dưỡng cơ Duchenne, đặc biệt ở trẻ nam;
- Triệu chứng yếu cơ nặng dần, kèm theo sốt, khó thở, mệt mỏi hoặc giảm khả năng vận động rõ rệt.
Phương pháp điều trị loạn dưỡng cơ Duchenne
Hiện nay, loạn dưỡng cơ Duchenne (DMD) vẫn chưa có phương pháp điều trị giúp chữa khỏi hoàn toàn. Tuy nhiên, nhiều nhóm thuốc và biện pháp hỗ trợ khác đã được chứng minh có khả năng làm chậm tiến triển bệnh, giảm triệu chứng, bảo vệ cơ tim – phổi và cải thiện chất lượng sống, từ đó giúp kéo dài tuổi thọ đáng kể cho người bệnh. Cụ thể:
Dùng thuốc
 Điều trị loạn dưỡng cơ Duchenne bằng thuốc
Điều trị loạn dưỡng cơ Duchenne bằng thuốc
Corticosteroid: Prednisone và deflazacort là hai loại thuốc được sử dụng phổ biến nhất. Các nghiên cứu cho thấy corticosteroid giúp làm chậm quá trình yếu cơ, duy trì khả năng vận động độc lập lâu hơn, đồng thời có thể giảm nguy cơ biến dạng cột sống và cải thiện chức năng hô hấp.
Thuốc chỉnh sửa exon (Exon-skipping): Phù hợp cho nhóm bệnh nhân có đột biến gene dystrophin nằm trong vùng có thể chỉnh sửa exon. Các thuốc như eteplirsen, golodirsen, viltolarsen hoặc casimersen được thiết kế để giúp cơ thể tạo ra dạng dystrophin ngắn hơn nhưng vẫn có chức năng một phần. Tuy vậy, hiệu quả của nhóm điều trị này còn hạn chế, chỉ áp dụng cho một số kiểu đột biến nhất định.
Thuốc điều trị tim mạch: Bao gồm thuốc ức chế men chuyển (ACE inhibitors) hoặc thuốc chẹn beta nhằm kiểm soát bệnh cơ tim – một biến chứng thường gặp ở trẻ mắc DMD. Điều trị sớm giúp làm chậm suy tim và kéo dài tuổi thọ.
Các bài tập phục hồi chức năng
Phục hồi chức năng là phần không thể thiếu trong quản lý DMD, giúp duy trì độ linh hoạt của khớp, giảm co rút và hỗ trợ vận động.
- Tập vật lý trị liệu với các bài tập vận động nhẹ nhàng.
- Kéo giãn cơ thường xuyên để ngăn ngừa tình trạng co rút gân và cứng khớp.
- Liệu pháp thủy trị liệu (tập dưới nước) giúp giảm trọng lực lên cơ, hỗ trợ vận động hiệu quả mà không gây quá tải.
- Tránh mọi hình thức tập luyện quá sức, vì có thể làm tổn thương cơ trầm trọng hơn.
Phẫu thuật
Phẫu thuật được xem xét trong một số trường hợp để cải thiện chức năng và chất lượng sống:
- Phẫu thuật gân hoặc khớp: nhằm giảm co rút, cải thiện tư thế đứng – ngồi và tăng khả năng vận động.
- Phẫu thuật chỉnh vẹo cột sống: thường được thực hiện khi trẻ đã chuyển sang giai đoạn phải dùng xe lăn, giúp giảm nguy cơ biến chứng hô hấp và cải thiện ổn định cột sống.
Kết luận
Loạn dưỡng cơ Duchenne là bệnh lý tiến triển nhanh, nhưng hoàn toàn có thể được quản lý hiệu quả nếu phát hiện sớm và áp dụng đúng phác đồ điều trị. Bên cạnh đó, phòng ngừa bằng tư vấn di truyền, xét nghiệm mang gene và sàng lọc trước sinh là giải pháp giúp giảm thiểu nguy cơ mắc bệnh ở thế hệ sau.






