Tìm hiểu chung rối loạn chuyển hoá Purine và Pyrimidine
Trước khi tìm hiểu chi tiết hơn về rối loạn chuyển hoá Purine và Pyrimidine là gì, chúng ta cần hiểu Purine và Pyrimidine được định nghĩa như thế nào:
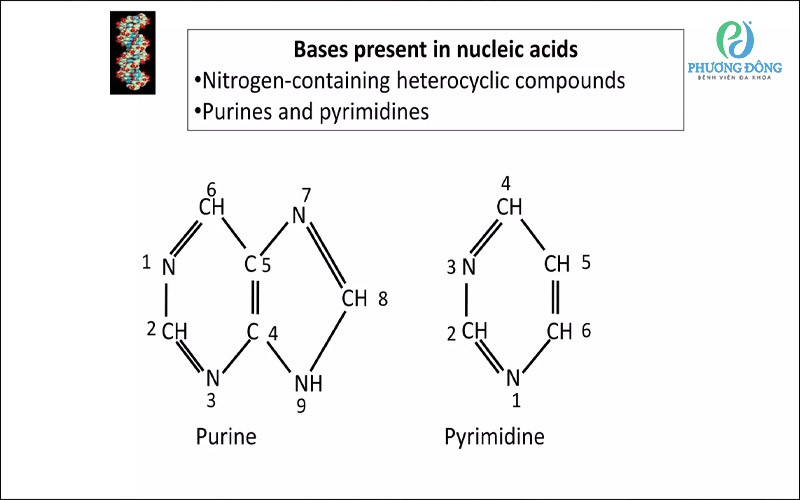 Nhận định Purine và Pyrimidine
Nhận định Purine và Pyrimidine
Purine là gì?
Purine là một lại hợp chất hữu cơ dị vòng thơm, có mặt tự nhiên trong cơ thể người và nhiều loại thực phẩm. Chúng giữ vai trò thiết yếu trong quá trình tổng hợp vật chất di truyền, cụ thể là DNA và RNA, giúp duy trì hoạt động sống của tế bào. Thuật ngữ “ Purine” được nhà hoá học Emil Fischer đặt ra vào năm 1884 và được tổng hợp nhân tạo lần đầu vào năm 1898, mở ra nền tảng cho nghiên cứu sinh học phân tử sau này.
Về mặt cấu trúc, purine gồm hai vòng hydrocarbon kết hợp với bốn nguyên tử nito, tạo nên các nucleobase quan trọng là adenine (A) và guanine (G). Hai base này là thành phần không thể thiếu trong cấu trúc của DNA và RNA, giúp lưu trữ và truyền đạt thông tin di truyền giữa các thế hệ tế bào.
Các dạng purine trong cơ thể con người
- Purine nội sinh: Được tự tổng hợp trong quá trình chuyển hoá acid nucleic của tế bào.
- Purine ngoại sinh: Được cung cấp qua thực phẩm và đồ uống, đặc biệt là thịt đỏ, nội tạng động vật, hải sản, rượu bia và một số loại đậu.
Quá trình chuyển hoá purine và hình thành acid uric
Khi cơ thể tiêu hoá và chuyển hoá purine, chúng sẽ tạo ra acid uric – một sản phẩm cuối cùng của quá trình này. Ở mức độ bình thường, acid uric như một chất chống oxy hoá, góp phần bảo vệ tế bào thần kinh và hỗ trợ hoạt động của não bộ.
Tuy nhiên, khi nồng độ acid uric trong máu tăng quá cao, cơ thể không thể đào thải hết qua thận, dẫn đến tích tụ tinh thể urat tại các khớp. Tình trạng này kích hoạt phản ứng viêm của hệ miễn dịch, gây ra các cơn đau gout cấp với triệu chứng sưng, nóng, đỏ, đau dữ dội ở khớp – thường gặp nhất ở ngón chân cái.
Pyrimidine là gì?
Pyrimidine là một hợp chất hữu cơ thuộc nhóm bazo nito (nitrogenous base), có vai trò nền tảng trong cấu trúc của DNA và RNA. Đây là thành phần tạo nên các nucleobase quan trọng gồm cytosine (C), thymine (T) và uracil (U) – 3 loại bazo tham gia trực tiếp vào quá trình mã hoá, sao chép và phiên mã vật chất di truyền.
Về mặt cấu trúc, pyrimidine có một vòng thơm gồm sau nguyên tử, trong đó có hai nguyên tử nitơ và bốn nguyên tử cacbon. Cấu trúc này giúp pyrimidine dễ dàng liên kết hydro với purine để tạo thành các cặp bazo ổn định trong DNA và RNA.
Lịch sử nghiên cứu về pyrimidine bắt đầu từ năm 1884, khi các nhà khoa học bắt đầu khám phá bản chất của hợp chất này. Đến năm 1885, nhà hoá học Pinner lần đầu tiên đặt ra thuật ngữ “pyrimidine”, mở ra nền tảng cho hàng loạt công trình nghiên cứu sâu hơn về cấu trúc và vai trò sinh học của nó.
Ngày nay, pyrimidine và các dẫn xuất của nó không chỉ được xem là thành phần cấu tạo của axit nucleic, mà còn là nguyên liệu quan trọng trong quá trình tổng hợp vitamin, axit amin và coenzyme. Nhờ đó, pyrimidine góp phần duy trì chuyển hoá năng lượng, tăng trưởng tế bào và hoạt động sinh lý bình thường của cơ thể.
Sự khác biệt giữa Purine và Pyrimidine là gì?
|
Purine
|
Pyrimidine
|
- Bao gồm các nucleobase như Adenine (A), Guanine (G).
- Có 2 vòng hydrocacbon
- Có 4 nguyên tử nitơ
- Sản phẩm cuối cùng là acid uric
- Quá trình tổng hợp phức tạp.
|
- Bao gồm các nucleobase như Thymine (T), Uracil (U), Cytosine (C)
- Chỉ có 1 vòng hydrocacbon
- Có 2 nguyên tử nitơ
- Sản phẩm cuối cùng là ure
- Quá trình tổng hợp đơn giản hơn.
|
Cả Purin và Pyrimidine đều là sản phẩm chuyển hoá của acid Nucleic trong cơ thể. Các rối loạn chuyển hóa Purin và Pyrinmidine sẽ dẫn đến sự thay đổi nồng độ sản phẩm chuyển hoá trung gian hoặc sản phẩm chuyển hoá cuối cùng của hai phân tử này.
Dấu hiệu và triệu chứng của rối loạn chuyển hoá Purine và Pyrimidine
Rối loạn chuyển hoá purine và pyrimidine là nhóm bệnh di truyền hiếm gặp, xảy ra khi cơ thể thiếu hoặc rối loạn hoạt động của các enzyme tham gia vào quá trình tổng hợp và phân giải các bazơ nitơ purine – pyrimidine. Những rối loạn này có thể ảnh hưởng nghiêm trọng đến hệ miễn dịch, cơ xương, thần kinh và chuyển hóa năng lượng.
Rối loạn chuyển hoá purine
Thiếu hụt enzyme Adenosine Deaminase (ADA deficiency)
Đây là rối loạn di truyền lặn trên nhiễm sắc thể thường, do gen ADA bị đột biến khiến cơ thể không thể chuyển hoá adenosine thành inosine. Hậu quả là adenine tích tụ, gây độc cho tế bào lympho và dẫn đến suy giảm miễn dịch kết hợp nặng (Severe Combined Immunodeficiency – SCID).
Triệu chứng:
- Thường khởi phát sớm ở giai đoạn sơ sinh: viêm phổi dai dẳng, tiêu chảy kéo dài, nhiễm trùng tái phát.
- Nếu không điều trị, trẻ có thể tử vong trong 1–2 năm đầu đời.
- Một số thể nhẹ hơn (thiếu hụt enzyme một phần) chỉ gây suy giảm miễn dịch mức độ nhẹ hoặc không có triệu chứng rõ ràng.
Điều trị: Ghép tuỷ xương hoặc ghép tế bào gốc máu cuống rốn là phương pháp điều trị đặc hiệu. Ngoài ra, liệu pháp gen ADA cũng đang được ứng dụng trong một số trung tâm lớn trên thế giới.
Thiếu hụt enzyme Myoadenylate Deaminase (MADA)
Enzyme này chủ yếu tồn tại trong cơ xương, có vai trò chuyển hoá adenosine monophosphate (AMP) thành inosine. Khi thiếu hụt enzyme, quá trình chuyển hoá năng lượng trong cơ bị gián đoạn.
Triệu chứng:
- Đau cơ, chuột rút, mỏi và yếu cơ sau vận động hoặc gắng sức.
- Một số bệnh nhân không có biểu hiện lâm sàng, chỉ phát hiện tình cờ khi làm xét nghiệm enzyme cơ.
Điều trị: Hiện chưa có thuốc đặc hiệu. Người bệnh được khuyến khích điều chỉnh cường độ luyện tập, nghỉ ngơi hợp lý và duy trì dinh dưỡng cân đối để giảm triệu chứng mệt cơ.
Thiếu hụt enzyme Purine Nucleoside Phosphorylase (PNP deficiency)
PNP là enzyme có mặt ở nhiều mô, đặc biệt trong tế bào lympho, giúp chuyển hóa inosine thành hypoxanthine và guanosine thành guanine. Thiếu hụt enzyme này làm tích tụ purine độc hại và gây tổn thương hệ miễn dịch.
Triệu chứng:
- Xuất hiện sớm trong năm đầu đời: nhiễm trùng tái diễn (viêm phổi, viêm xoang, tiêu chảy kéo dài).
- Khoảng 70% bệnh nhân có biểu hiện thần kinh như: chậm phát triển trí tuệ, rối loạn vận động, mất thăng bằng, co cứng cơ.
Điều trị: Ghép tuỷ hoặc ghép tế bào gốc cuống rốn là phương pháp điều trị chủ yếu. Trong một số trường hợp, có thể áp dụng liệu pháp gen đang được nghiên cứu để phục hồi hoạt tính enzyme.
Tăng hoạt enzyme Phosphoribosylpyrophosphate (PRPP) Synthetase
Đây là bệnh di truyền lặn liên kết nhiễm sắc thể X, khiến enzyme PRPP synthetase hoạt động quá mức, dẫn đến tăng tổng hợp purine và hậu quả là tăng acid uric trong máu và nước tiểu.
Triệu chứng:
- Thể nhẹ: xuất hiện tinh thể urat trong nước tiểu, đau khớp tái diễn do lắng đọng tinh thể urat (dấu hiệu sớm của bệnh gout).
- Thể nặng: giảm thính lực, giảm trương lực cơ, rối loạn vận động và mất phối hợp động tác.
Điều trị: Kiểm soát nồng độ acid uric bằng chế độ ăn ít purine, tránh rượu bia và thuốc làm tăng acid uric. Có thể sử dụng thuốc ức chế xanthine oxidase (như allopurinol) để ngăn ngừa gout và tổn thương thận.
Rối loạn chuyển hoá pyrimidine
Rối loạn này chủ yếu do thiếu hụt enzyme Uridine Monophosphate Synthase (UMPS) – một enzyme cần thiết trong quá trình tổng hợp pyrimidine. Khi thiếu hụt, cơ thể tăng đào thải acid orotic (urotic acid), dẫn đến ứ đọng trong máu và nước tiểu.
Triệu chứng lâm sàng:
- Thiếu máu nguyên bào khổng lồ do giảm tổng hợp DNA trong tủy xương.
- Rối loạn thần kinh: chậm phát triển thể chất, thiểu năng trí tuệ, co giật hoặc động kinh.
- Nước tiểu đục do tinh thể acid orotic; trong trường hợp nặng có thể gây tắc nghẽn niệu đạo hoặc trào ngược bàng quang – niệu quản dẫn đến tiểu ra máu.
- Một số trường hợp kèm dị tật bẩm sinh như thông liên thất, lác mắt hoặc loãng xương.
Điều trị: Bổ sung uridine giúp ức chế sản xuất orotic acid và cải thiện thiếu máu và theo dõi chức năng thận định kỳ để phòng ngừa biến chứng tiết niệu.
Xem thêm:
Chẩn đoán và điều trị rối loạn chuyển hoá Purine và Pyrimidine
Rối loạn chuyển hoá Purine và Pyrimidine là bệnh lý cực kỳ hiếm, với tỷ lệ ước tính khoảng 1/1.000.000 trẻ sơ sinh. Tính đến nay, y văn thế giới chỉ ghi nhận khoảng 20 trường hợp được chẩn đoán xác định. Bệnh di truyền theo kiểu lặn trên nhiễm sắc thể thường, nghĩa là trẻ chỉ mắc bệnh khi nhận hai gen đột biến từ cả cha và mẹ.
Chẩn đoán
Việc chẩn đoán dựa trên sự kết hợp giữa biểu hiện lâm sàng và xét nghiệm cận lâm sàng chuyên sâu.
Các triệu chứng thường gặp gồm: thiếu máu hồng cầu to kháng trị với acid folic và vitamin B12, chậm phát triển thể chất, mệt mỏi kéo dài và có thể xuất hiện tinh thể acid orotic trong nước tiểu.
 Xét nghiệm máu giúp hỗ trợ xác định tình trạng bệnh
Xét nghiệm máu giúp hỗ trợ xác định tình trạng bệnh
- Xét nghiệm nước tiểu: Cho thấy nồng độ acid orotic tăng cao bất thường, là dấu hiệu đặc trưng của bệnh.
- Xét nghiệm máu: Phát hiện giảm hồng cầu, tăng MCV (thể tích trung bình hồng cầu) và có thể thấy rối loạn chuyển hóa pyrimidine.
- Chẩn đoán xác định: Dựa vào xét nghiệm di truyền phân tử, phát hiện đột biến gen UMPS – gen chịu trách nhiệm mã hóa enzyme uridine monophosphate synthase. Đây là kỹ thuật phức tạp, chỉ được thực hiện tại một số ít phòng xét nghiệm di truyền chuyên sâu trên thế giới.
Điều trị
Hiện nay, uridine triacetate (Xuriden) là thuốc điều trị đặc hiệu duy nhất được Cục Quản lý Thực phẩm và Dược phẩm Hoa Kỳ (FDA) phê duyệt vào năm 2015 dành cho bệnh orotic niệu di truyền.
Bản chất của Xuriden là tiền chất của uridine monophosphate (UMP) – hợp chất mà bệnh nhân bị thiếu hụt do khiếm khuyết enzyme UMPS. Khi được bổ sung, thuốc giúp:
- Bù đắp sự thiếu hụt pyrimidine nội sinh, khôi phục quá trình tổng hợp ADN và ARN bình thường.
- Cải thiện tình trạng thiếu máu hồng cầu to, giúp tăng sinh hồng cầu hiệu quả.
- Giảm nồng độ acid orotic trong nước tiểu, hạn chế tổn thương thận và rối loạn chuyển hoá thứ phát.
- Hỗ trợ tăng trưởng và phát triển thể chất, đặc biệt ở trẻ em mắc bệnh.
Điều trị bằng uridine triacetate cần được duy trì suốt đời, với liều lượng và thời gian sử dụng do bác sĩ chuyên khoa chuyển hóa hoặc di truyền chỉ định. Việc ngưng thuốc hoặc điều trị không liên tục có thể khiến các triệu chứng tái phát, gây ảnh hưởng nghiêm trọng đến sức khỏe và sự phát triển toàn thân của người bệnh.
Theo dõi và quản lý lâu dài
Người bệnh cần được theo dõi định kỳ để đánh giá chức năng gan, thận, huyết học và phát triển thể chất. Ngoài ra, tư vấn di truyền cho gia đình là bước cần thiết để phát hiện sớm người mang gen bệnh và hướng dẫn kế hoạch sinh sản an toàn.
Kết luận
Rối loạn chuyển hoá Purine và Pyrimidine là nhóm bệnh ít được chú ý nhưng hậu quả lại không hề nhỏ, ảnh hưởng đến nhiều cơ quan quan trọng như gan, thận, thần kinh và hệ cơ xương. Việc phát hiện sớm thông qua xét nghiệm sinh hoá, kiểm tra enzyme và định lượng acid uric có vai trò quyết định trong chẩn đoán và điều trị. Chủ động tầm soát và xây dựng chế độ dinh dưỡng chính là giải pháp để bảo vệ sức khỏe bản thân.




")

