Bệnh Fabry là một dạng rối loạn di truyền hiếm gặp nhưng lại gây tổn thương nghiêm trọng lên nhiều cơ quan quan trọng như tim, thận, hệ thần kinh và mạch máu. Do bệnh tiến triển âm thầm trong nhiều năm nên nhiều người chỉ phát hiện bệnh khi cơ thể đã bị tổn thương nặng, thậm chí là đối mặt với nguy cơ đột quỵ, suy thận hoặc thậm chí là tử vong – vô cùng nguy hiểm.
Bệnh Fabry là bệnh gì? Tỷ lệ mắc bệnh và mức độ phổ biến
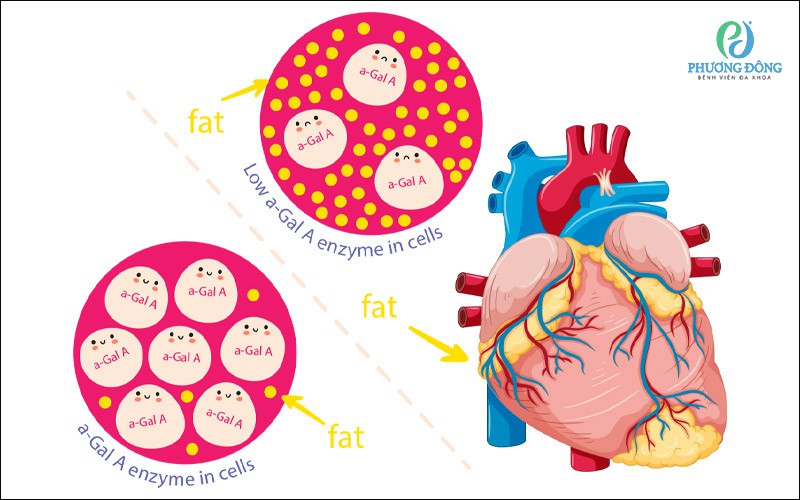 Bệnh Fabry là do sự thiếu hụt enzyme alpha-galactosidase A
Bệnh Fabry là do sự thiếu hụt enzyme alpha-galactosidase A
Bệnh Fabry, hay còn gọi là Anderson–Fabry, là một rối loạn di truyền hiếm thuộc nhóm bệnh dự trữ lysosome. Nguyên nhân chính của bệnh là do đột biến gen GLA, khiến cơ thể thiếu hụt hoặc giảm chức năng enzyme alpha-galactosidase A (α-GAL A). Đây là enzyme có vai trò phân giải globotrialosylceramide (Gb3 hay GL-3) – một loại chất béo cần được loại bỏ khỏi tế bào trong quá trình chuyển hóa bình thường.
Khi enzyme α-GAL A hoạt động không đầy đủ, Gb3 tích tụ dần trong nhiều loại mô và cơ quan khác nhau, đặc biệt là mạch máu, tim, thận, hệ thần kinh và da. Sự tích tụ kéo dài này làm ảnh hưởng tới quá trình hoạt động của tế bào và dẫn đến nhiều biểu hiện lâm sàng đa dạng, có thể khởi phát từ thời thơ ấu và tiến triển nặng theo thời gian.
Mặc dù xuất hiện ở mọi chủng tộc, bệnh Fabry được xếp vào nhóm bệnh hiếm, do đó việc xác định chính xác mức độ phổ biến gặp nhiều khó khăn. Tuy nhiên, các nghiên cứu dịch tễ học hiện đại cho thấy tần suất mắc bệnh có thể cao hơn nhiều so với ước tính trước đây, dao động từ 1/1.400 đến 1/9.000 người trong một số quần thể nhất định. Điều này cho thấy dù Fabry là bệnh hiếm, nhưng mức độ hiện diện trong cộng đồng có thể phổ biến hơn so với nhận định truyền thống.
Ai có nguy cơ mắc bệnh Fabry?
Bệnh Fabry xảy ra khi một cá nhân thừa hưởng gen GLA bị đột biến trên nhiễm sắc thể X từ cha hoặc mẹ. Vì cơ chế di truyền liên quan đến nhiễm sắc thể X, khả năng mắc bệnh hoặc mang gen đột biến sẽ khác nhau giữa nam và nữ. Nam giới chỉ có một nhiễm sắc thể X từ mẹ, trong khi nữ giới có hai nhiễm sắc thể X, được thừa hưởng từ cả bố và mẹ.
Cha mẹ có thể truyền gen đột biến gây bệnh Fabry cho con theo các cách sau:
- Bố mắc bệnh Fabry sẽ truyền toàn bộ nhiễm sắc thể X mang gen lỗi cho tất cả con gái, khiến tất cả các bé gái đều mang đột biến liên quan đến bệnh Fabry. Ngược lại, con trai của bố mắc bệnh không bị ảnh hưởng, vì bé trai nhận nhiễm sắc thể Y từ bố thay vì nhiễm sắc thể X.
- Mẹ mang gen hoặc mắc bệnh Fabry có 50% khả năng truyền nhiễm sắc thể X bị đột biến cho cả con trai và con gái. Vì vậy, mỗi đứa trẻ sinh ra đều có khả năng thừa hưởng gen lỗi hoặc không, tùy thuộc vào nhiễm sắc thể X mà trẻ nhận được.
Nguyên nhân gây bệnh Fabry
Đột biến tại gen galactosidase alpha (GLA) là nguyên nhân trực tiếp dẫn đến bệnh Fabry. Gen này chịu trách nhiệm mã hóa enzyme alpha-galactosidase A (α-GAL A) – một loại enzyme tham gia vào quá trình phân hủy nhóm chất béo thuộc sphingolipid trong lysosome. Khi gen GLA bị đột biến, cơ thể không thể sản xuất đủ hoặc hoàn toàn thiếu enzyme α-GAL A.
Hậu quả là các phân tử chất béo không được phân hủy sẽ tích tụ dần trong thành mạch máu, tế bào thần kinh, tim, thận và nhiều cơ quan khác, từ đó gây tổn thương đa cơ quan và dẫn đến các biểu hiện đặc trưng của bệnh Fabry.
Dấu hiệu của bệnh Fabry
Các triệu chứng của bệnh Fabry có thể rất đa dạng và xuất hiện ở nhiều độ tuổi khác nhau. Các dấu hiệu và triệu chứng phổ biến bao gồm:
- Đau và cảm giác nóng rát ở tay và chân (acroparesthesia)
- Phát ban da (u mạch sừng hoá) xuất hiện dưới dạng đốm nhỏ màu đỏ sẫm;
- Mệt mỏi và yếu ớt;
- Giảm tiết mồ hôi;
- Các vấn đề về đường tiêu hoá;
- Rối loạn chức năng thận và suy thận;
- Bất thường về tim, chẳng hạn như bệnh cơ tim phì đại;
- Mất thính lực và ù tai;
- Các vấn đề về thị lực;
- U mạch sừng hoá (những đốm nhỏ, không phải ung thư, màu đỏ tím nổi trên da) có thể phát triển ở phần dưới của thân và trở nên nhiều hơn theo tuổi tác.
 Phát ban da dạng mảng màu đỏ hoặc tím, thường xuất hiện ở vùng lưng hoặc háng người bệnh
Phát ban da dạng mảng màu đỏ hoặc tím, thường xuất hiện ở vùng lưng hoặc háng người bệnh
Biến chứng của bệnh Fabry
Sự tích tụ chất béo trong nhiều năm có thể làm hỏng mạch máu và dẫn đến các vấn đề đe doạ tính mạng, bao gồm:
- Các vấn đề về tim, bao gồm loạn nhịp tim, đau tim, phì đại tim và suy tim;
- Suy thận;
- Tổn thương thần kinh (bệnh lý thần kinh ngoại biên);
- Đột quỵ, bao gồm cả các cơn thiếu máu não thoáng qua (TIA hoặc đột quỵ nhẹ).
Xem thêm:
Chẩn đoán và điều trị bệnh Fabry
Để chẩn đoán bệnh Fabry, bác sĩ sẽ chỉ định một số xét nghiệm nhằm đánh giá hoạt động enzyme và xác định bất thường di truyền. Các kỹ thuật được sử dụng phổ biến gồm:
Xét nghiệm enzyme: Đây là phương pháp đo hoạt độ enzyme alpha-galactosidase A (α-GAL A) trong máu. Khi mức enzyme giảm xuống dưới 1% so với bình thường, khả năng cao bệnh nhân mắc bệnh Fabry. Tuy nhiên, xét nghiệm này chỉ có độ tin cậy cao ở nam giới, vì nữ giới mang gen bệnh có thể có hoạt độ enzyme bình thường hoặc dao động.
Xét nghiệm di truyền: Do nhiều trường hợp nam giới mắc bệnh vẫn có hoạt độ enzyme gần mức bình thường, xét nghiệm giải trình tự gen GLA được sử dụng để xác nhận chính xác đột biến gây bệnh. Đối với nữ giới, xét nghiệm di truyền là tiêu chuẩn vàng để chẩn đoán.
Sinh thiết: Một số trường hợp bác sĩ có thể chỉ định sinh thiết da hoặc thận để tìm các thể vùi đặc trưng trong tế bào, hỗ trợ xác định tổn thương do Fabry gây ra.
Chẩn đoán hình ảnh: Nếu bệnh nhân có dấu hiệu thần kinh như nghi ngờ nhồi máu não hoặc đột quỵ, bác sĩ sẽ chỉ định chụp cộng hưởng từ (MRI não) để phát hiện tổn thương mạch máu hoặc các biến chứng liên quan.
Hiện nay, dù chưa có phương pháp điều trị giúp chữa khỏi hoàn toàn bệnh Fabry, nhưng nhiều lựa chọn điều trị hiện đại có thể làm giảm triệu chứng, làm chậm tiến triển bệnh và cải thiện chất lượng cuộc sống. Những phương pháp điều trị chính gồm:
Liệu pháp thay thế enzyme (ERT): Agalsidase beta và Agalsidase alfa là hai chế phẩm enzyme tổng hợp được dùng để thay thế enzyme α-GAL A bị thiếu hụt. Chúng hoạt động tương tự enzyme tự nhiên, giúp giảm tích tụ globotriaosylceramide (Gb3) trong tế bào – nguyên nhân chính gây bệnh Fabry. Liệu pháp được thực hiện bằng truyền tĩnh mạch định kỳ, thường mỗi 2 tuần.
Điều trị nội khoa: Một số thuốc được kê đơn nhằm kiểm soát triệu chứng và hạn chế biến chứng của bệnh, bao gồm:
- Thuốc chống co giật (Carbamazepine, Phenytoin) giúp giảm đau kiểu thần kinh ở tay và chân.
- Thuốc điều trị rối loạn tiêu hoá như Metoclopramide cho triệu chứng buồn nôn hoặc chậm tiêu.
- Thuốc ổn định enzyme α-GAL A như Migalastat, dùng cho bệnh nhân mang đột biến gen “nhạy” với thuốc, giúp tối ưu hoạt động của enzyme còn lại trong cơ thể.
Các phương pháp hỗ trợ khác: Trong giai đoạn muộn khi thận bị tổn thương nặng, bệnh nhân có thể cần lọc máu hoặc ghép thận để duy trì chức năng cơ quan. Đây là giải pháp quan trọng giúp kéo dài thời gian sống và cải thiện sức khỏe tổng thể.
Kết luận
Fabry không phải là một căn bệnh phổ biến, nhưng mức độ nguy hiểm của nó lại vượt xa nhiều bệnh lý mạn tính do khả năng phá huỷ âm thầm trong thời gian dài. Việc nhận biết sớm các triệu chứng, và điều trị đúng hướng có thể giúp người bệnh kiểm soát sự tiến triển của bệnh, giảm nguy cơ biến chứng. Nếu bạn hoặc người thân có biểu hiện nghi ngờ mắc bệnh Fabry, hãy chủ động thăm khám để được chẩn đoán kịp thời.






