Hội chứng Hurler là một dạng rối loạn chuyển hóa bẩm sinh hiếm gặp nhưng đặc biệt nguy hiểm, khi cơ thể trẻ không thể phân hủy các chất mucopolysaccharide, khiến độc chất tích tụ dần trong tế bào và làm tổn thương nhiều cơ quan. Bệnh tiến triển nhanh, ảnh hưởng xương khớp, thị lực, tim mạch và cả khả năng phát triển trí tuệ, khiến việc chẩn đoán và can thiệp sớm trở nên vô cùng quan trọng.
Hội chứng Hurler là gì?
Hội chứng Hurler là thể bệnh nặng nhất trong nhóm mucopolysaccharidosis type 1 (MPS I) – một rối loạn di truyền lặn trên nhiễm sắc thể thường. Bệnh xảy ra khi cơ thể không sản xuất đủ enzyme alpha-L-iduronidase, dẫn đến tích tụ các phân tử đường phức tạp bên trong tế bào.
MPS I xuất hiện khi cơ thể không đủ enzyme để phân giải nhóm phân tử đường glycosaminoglycan (GAGs) – trước đây được gọi là mucopolysaccharides. Khi GAGs tích tụ, chúng gây tổn thương nhiều cơ quan và dẫn đến các biểu hiện lâm sàng nghiêm trọng, bao gồm:
- Biến dạng xương và các vấn đề khớp
- Đặc điểm khuôn mặt thô, không điển hình
- Suy giảm phát triển nhận thức và vận động
- Bệnh lý tim mạch và rối loạn hô hấp
- Gan và lách to do tích tụ chất trong tế bào
Hội chứng Hurler thuộc nhóm bệnh tích tụ thể lysosome. Ở người khỏe mạnh, lysosome đóng vai trò “nhà máy xử lý chất thải” của tế bào – giúp lưu trữ, tái chế và phân giải các phân tử. Khi enzyme bị thiếu hụt, các phân tử không được phân hủy sẽ tích tụ trong lysosome. Lượng chất tồn dư tăng cao khiến tế bào suy yếu, hoạt động bất thường hoặc chết sớm, từ đó gây ra hàng loạt triệu chứng của bệnh.
Trẻ mắc hội chứng Hurler thường có tuổi thọ ngắn, chủ yếu do các biến chứng đe dọa tính mạng như suy tim, nhiễm trùng hô hấp tái phát hoặc suy đa cơ quan nếu không được điều trị sớm và đúng cách.
Đối tượng có nguy cơ mắc hội chứng Hurler
Hội chứng Hurler có thể xuất hiện ở bất kỳ trẻ nào vì đây là bệnh lý di truyền lặn trên nhiễm sắc thể thường, bắt nguồn từ đột biến ngẫu nhiên trên gen IDUA. Tuy nhiên, nguy cơ mắc bệnh sẽ cao hơn nếu trong gia đình đã từng có người bị mucopolysaccharidosis type I (MPS I), bao gồm cả thể Hurler, Hurler–Scheie hoặc Scheie. Trong trường hợp cha mẹ đều mang gen lặn gây bệnh, khả năng sinh con mắc hội chứng Hurler tăng đáng kể.
Triệu chứng của hội chứng Hurler
Các biểu hiện lâm sàng của hội chứng Hurler rất đa dạng và mức độ nặng nhẹ có thể khác nhau giữa từng trẻ. Các triệu chứng thường khởi phát từ giai đoạn đầu đời, tiến triển rõ rệt trong thời thơ ấu và kéo dài đến tuổi vị thành niên nếu không được điều trị kịp thời.
Một dấu hiệu đặc trưng giúp phân biệt hội chứng Hurler với các thể nhẹ hơn của bệnh MPS I là tình trạng chậm phát triển trí tuệ, kèm suy giảm dần khả năng ghi nhớ và tiếp thu ở trẻ. Ở các thể MPS I nhẹ hơn, trí tuệ thường không bị ảnh hưởng đáng kể.
Các triệu chứng thường gặp của hội chứng Hurler có thể bao gồm:
- Các bệnh lý về tim, đặc biệt là tổn thương van tim hoặc bệnh cơ tim.
- Giảm thính lực tiến triển do tích tụ mucopolysaccharide trong tai giữa và tai trong.
- Tích tụ dịch não tủy quanh não, dẫn đến não úng thủy.
- Phì đại các cơ quan như gan, lách, amidan, mô liên kết hoặc cơ.
- Các rối loạn về thị giác, bao gồm tăng nhãn áp hoặc đục giác mạc.
- Các vấn đề cơ – xương – khớp như căng cơ, hội chứng ống cổ tay và bệnh lý khớp khiến vận động hạn chế.
- Nhiễm trùng hô hấp tái phát, ngưng thở khi ngủ, khó thở do đường thở hẹp.
- Thoát vị rốn hoặc thoát vị bẹn.
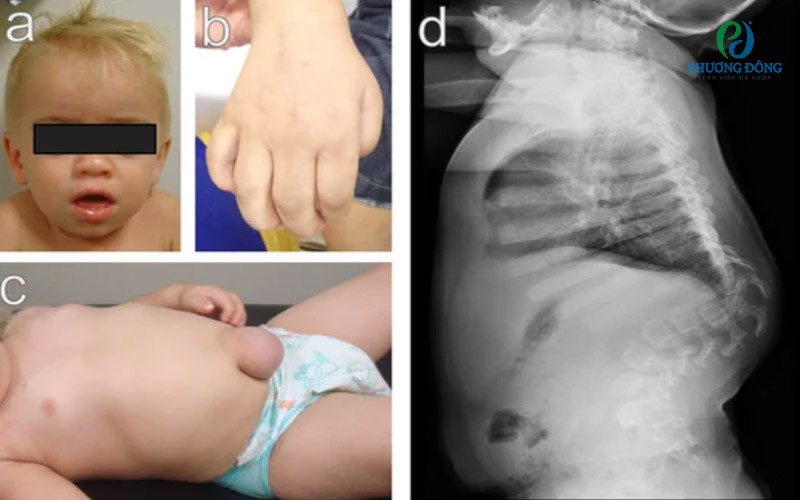 Các đặc điểm của hội chứng Hurler.
Các đặc điểm của hội chứng Hurler.
Ghi chú:
- (a). Các đặc điểm khuôn mặt thô. Lưu ý trán rộng, cổ ngắn, đầu mũi rộng, các đặc điểm khuôn mặt thô và đục giác mạc.
- (b). Bàn tay vuốt do ngón tay bị cong vẹo.
- (c). Thoát vị rốn.
- (d). Nhìn nghiêng cột sống. Lưu ý gù cột sống ngực-thắt lưng và xương sườn rộng. Đã có sự đồng ý của bệnh nhân cho việc sử dụng hình ảnh.
Đặc điểm vật lý
Ngay trong năm đầu đời, nhiều trẻ mắc hội chứng Hurler đã xuất hiện các đặc điểm hình thái đặc trưng. Những dấu hiệu này thường tiến triển theo thời gian, bao gồm:
- Thấp bé so với độ tuổi do rối loạn phát triển xương.
- Loạn cốt hóa – xương phát triển bất thường, gây biến dạng khung xương.
- Gù lưng vùng ngực – thắt lưng (kyphosis) do bất thường cột sống.
- Rậm lông nhiều hơn so với trẻ khỏe mạnh cùng độ tuổi.
Xem thêm:
Nguyên nhân gây ra hội chứng Hurler
Hội chứng Hurler chủ yếu bắt nguồn từ đột biến gen IDUA – gen chịu trách nhiệm mã hóa enzyme α-L-iduronidase. Đây là enzyme nằm trong lysosome, có vai trò phân giải các chất mucopolysaccharide (GAGs) – những phân tử cần được loại bỏ sau khi tế bào sử dụng.
Khi gen IDUA bị đột biến, cơ thể không sản xuất đủ enzyme α-L-iduronidase, khiến quá trình phân hủy GAGs bị đình trệ. Lúc này, các chất thải này bắt đầu tích tụ dần trong lysosome, làm tế bào phình to, rối loạn cấu trúc và giảm khả năng hoạt động.
Theo thời gian, sự tích tụ bất thường này làm tổn thương nhiều mô và cơ quan trong cơ thể. Khi tế bào không thể tự loại bỏ “rác thải” tích tụ, các biểu hiện lâm sàng đặc trưng của hội chứng Hurler sẽ xuất hiện và tiến triển ngày càng nặng hơn.
Hội chứng Hurler di truyền thế nào?
 Hội chứng Hurler di truyền theo kiểu lặn trên nhiễm sắc thể thường
Hội chứng Hurler di truyền theo kiểu lặn trên nhiễm sắc thể thường
Hội chứng Hurler là một rối loạn di truyền lặn trên nhiễm sắc thể thường, nghĩa là bệnh có thể được truyền từ cha mẹ sang con. Điều quan trọng cần hiểu là tình trạng này không phải do bất kỳ hành động nào của cha hoặc mẹ trong quá trình mang thai gây ra, mà xuất phát từ sai lệch trong vật chất di truyền.
Các tế bào sinh sản của cha mẹ (tinh trùng và trứng) đều mang một nửa bộ DNA của cơ thể. Khi hai tế bào này kết hợp, chúng tạo ra một tế bào mới với bộ gen hoàn chỉnh. Trong quá trình hình thành và phân chia tế bào, DNA phải được nhân đôi liên tục. Chính trong quá trình sao chép này, đột biến gen có thể xảy ra một cách ngẫu nhiên.
Khi xảy ra một “lỗi sao chép” trong DNA (đột biến), đoạn mã di truyền tại gen IDUA – gen chịu trách nhiệm sản xuất enzyme alpha-L-iduronidase – có thể bị thiếu hoặc sai lệch. Hậu quả là:
- Các tế bào không nhận được đầy đủ “hướng dẫn” cần thiết để hoạt động đúng chức năng.
- Enzyme alpha-L-iduronidase bị thiếu hụt hoặc không hoạt động.
- Chất mucopolysaccharides tích tụ trong tế bào, dẫn đến các biểu hiện của hội chứng Hurler.
Nói cách khác, đột biến xảy ra ở gen IDUA trong tế bào sinh sản của cha mẹ, và khi trẻ nhận phải hai bản sao gen lỗi (mỗi bản từ một phụ huynh), bệnh sẽ xuất hiện.
Chẩn đoán và điều trị hội chứng Hurler
Các xét nghiệm sàng lọc trước sinh như chọc ối hoặc sinh thiết có thể giúp phát hiện hội chứng Hurler ngay từ khi thai kỳ còn sớm. Hai phương pháp này cho phép kiểm tra những bất thường di truyền trong DNA của thai nhi, từ đó xác định nguy cơ mắc mucopolysaccharidosis type I (MPS I) – thể Hurler.
Sau khi trẻ chào đời, bác sĩ sẽ tiến hành khám lâm sàng để tìm kiếm những biểu hiện đặc trưng của bệnh và thực hiện xét nghiệm hoạt tính enzym nhằm khẳng định chẩn đoán. Việc khai thác tiền sử gia đình cũng rất quan trọng, vì MPS I là bệnh di truyền lặn trên nhiễm sắc thể thường, có thể gặp ở nhiều thành viên trong gia đình.
Để hỗ trợ chẩn đoán chính xác, trẻ có thể được yêu cầu thực hiện thêm các xét nghiệm chuyên sâu như X-quang đánh giá xương, siêu âm cơ quan, cùng với xét nghiệm máu và nước tiểu nhằm phát hiện sự tích tụ của glycosaminoglycans (GAGs).
Việc điều trị hội chứng Hurler chủ yếu tập trung vào kiểm soát triệu chứng, làm chậm tiến triển và ngăn ngừa biến chứng:
Liệu pháp thay thế enzyme (ERT): Bác sĩ sẽ chỉ định tiêm tĩnh mạch enzyme alpha-L-iduronidase (Aldurazyme) nhằm bù đắp lượng enzyme bị thiếu hụt. ERT có thể giúp cải thiện chức năng hô hấp, vận động, giảm tình trạng gan to và hạn chế tiến triển bệnh. Phác đồ tiêm thường được duy trì dài hạn, và tần suất điều trị sẽ được điều chỉnh dựa trên mức độ nặng của bệnh.
Ghép tế bào gốc tạo máu (HSCT): HSCT được xem là lựa chọn ưu tiên cho trẻ dưới 2 tuổi, hoặc những trẻ lớn hơn khi được bác sĩ đánh giá phù hợp. Phương pháp này có khả năng cải thiện tuổi thọ, làm chậm biến chứng thần kinh, duy trì chức năng nhận thức và giảm các biểu hiện tổn thương cơ thể. Tế bào gốc từ người hiến tặng mang enzyme khỏe mạnh sẽ thay thế tế bào khiếm khuyết của trẻ sau khi điều trị bằng hoá trị.
Các phương pháp điều trị khác có thể được áp dụng tùy theo triệu chứng của trẻ, bao gồm:
- Phẫu thuật: xử lý các biến chứng như thay van tim, ghép giác mạc, chỉnh hình xương hoặc chữa thoát vị.
- Vật lý trị liệu – trị liệu nghề nghiệp – trị liệu ngôn ngữ nhằm cải thiện vận động, giao tiếp và chất lượng cuộc sống.
- Hỗ trợ hô hấp bằng CPAP khi trẻ gặp khó thở hoặc ngưng thở khi ngủ.
- Sử dụng máy trợ thính cho trẻ có suy giảm thính lực.
- Dùng thuốc giảm đau để giảm bớt khó chịu liên quan đến các biến chứng xương – khớp và mô mềm.
Kết luận
Hội chứng Hurler là bệnh lý di truyền hiếm nhưng có tốc độ tiến triển nhanh và để lại hậu quả toàn thân nếu không được phát hiện kịp thời. Việc nắm rõ khái quát về bệnh và hiểu đúng các phương pháp điều trị giúp tối ưu hiệu quả điều trị và cải thiện chất lượng sống cho trẻ. Thăm khám sớm, tuân thủ phác đồ và theo dõi định kỳ với bác sĩ chuyên khoa là chìa khóa quan trọng nhất giúp kiểm soát bệnh và hạn chế biến chứng lâu dài.



 - Những thông tin cần biết")


